Switching to the enemy’s side: how a Huntington’s disease protein turns good cellular proteins bad
Scientists of the European Research Institute for the Biology of Ageing discovered that neurodegenerative disease-associated proteins hijack normal cellular proteins to promote their own aggregation and toxicity. The research by Olga Sin and others resulted from the collaboration between the research groups of Victor Guryev and Ellen Nollen. On March 16, the research article “Identification of an RNA Polymerase III Regulator Linked to Disease-Associated Protein Aggregation” was published in Molecular Cell (http://www.cell.com/molecular-cell/fulltext/S1097-2765(17)30137-5) and featured as the Editor’s choice in Science Signalling (http://stke.sciencemag.org/content/10/472/eaan3006.full).
In neurodegenerative diseases such as Alzheimer’s, Parkinson’s and Huntington’s disease, disease proteins aggregate in the brain and this has been linked to the loss of neurons. However, it is not clear which cellular mechanisms contribute to protein aggregation and how this is associated with neurodegeneration. In this study, the scientists set out to identify genes that promote protein aggregation during disease.
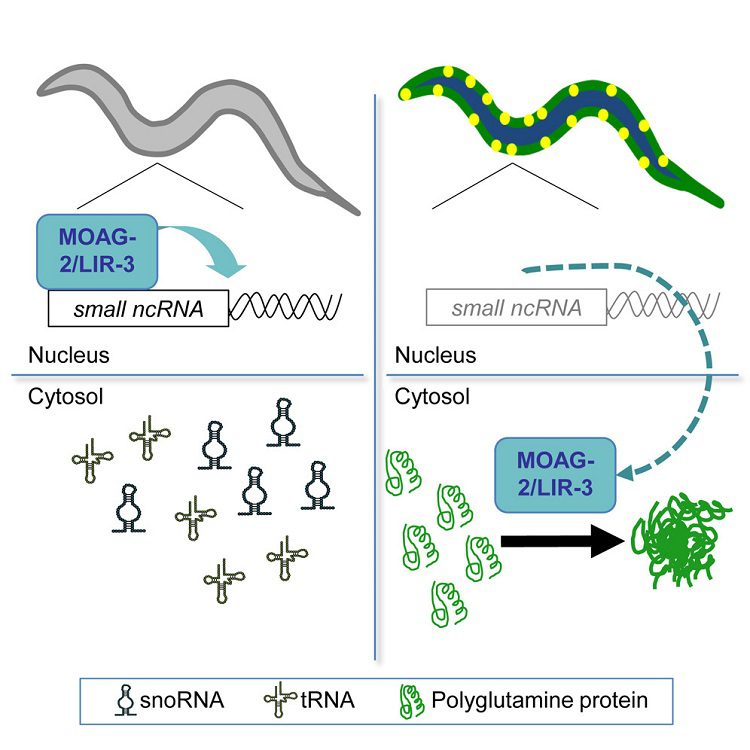
Using the roundworm Caenorhabditis elegans as a model for Huntington’s disease, the scientists performed a genetic screen to find genes that promote protein aggregation. The disruption of a gene called moag-2 (modifier of aggregation 2) was found to reduce disease-associated protein aggregation. The scientists discovered that MOAG-2 cooperates with the RNA Polymerase III complex to transcribe small non-coding RNAs. Surprisingly, this specific function of MOAG-2 was not related to the protein aggregation observed in the Huntington’s disease model. Instead, it was found that the Huntington’s disease protein relocates MOAG-2 from the nucleus to the cytoplasm and uses it to promote its own aggregation and toxicity in the cytoplasm.
This study reveals important insights on how neurodegenerative disease proteins alter cellular homeostasis that could explain what happens in the brains of affected patients. By interfering with protein trafficking between the nucleus and cytoplasm, the Huntington’s disease protein hijacks normal cellular proteins and uses them as “catalysts” to promote its own toxic protein aggregation. This cellular mechanism could be explored to develop disease-modifying treatments for Huntington’s disease in the future.